有没有ähå…Ïx³¨˜q™ä¸ªè¡Œä¸šçš„ç—›æ ¹å‘¢åQŸåˆ°æ—¶æ˜¯ä»€ä¹ˆï¼Œå¦‚何解决ã€?img src ="http://www.aygfsteel.com/rain1102/aggbug/419451.html" width = "1" height = "1" />
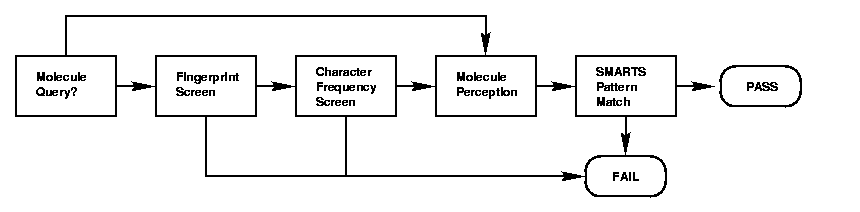
其大概æ„æ€å°±æ˜¯å…ˆé€šè¿‡Fingerprint˜q›è¡Œ½{›é€‰ï¼Œ˜q™æ ·å¯ä»¥å¿«é€Ÿçš„½{›é€‰æŽ‰ä¸€éƒ¨åˆ†æ•°æ®åQŒå¯¹äºŽå¤æ‚结构更有效åQ›å¦å¤–å°±æ˜¯æ ¹æ®åŽŸå个数或者特ŒDŠåŽŸå个数进行比较,如果查询¾l“构包å«ä¸‰ä¸ª“N”原ååQŒé‚£ä¹ˆæ‰€è¦æŸ¥è¯¢å‡ºçš„结构所å«æœ‰“N”的个数必™åÕd¤§äºŽç‰äº?åQŒè¿™æ ·å¯¹äºŽåŒ…å«ä¸€äº›ç‰¹ŒDŠå…ƒç´ 的效果是特别的好;˜q˜æœ‰ž®±æ˜¯æ ÒŽ(gu¨©)®åˆ†å的一些性质˜q›è¡Œ½{›é€‰è¿‡æ»¤ï¼Œæ¯”如芳香性ç‰åQ›æœ€åŽå†˜q›è¡ŒåŒšw…åQŒè¿™æ ·ä¸€æ¥å¯¹äºŽå¤æ‚结构以å?qi¨¢ng)å«ç‰Ò?gu¨©)®Šå…ƒç´ 的查询速度ä¼?x¨¬)æ高很多ã€?br /> 最åŽæ–‡ç« 丘q˜ç»™å‡ºæµ‹è¯•æ•°æ®ï¼Œä»Žä¸å¯ä»¥çœ‹å‡ºåQŒé€Ÿåº¦ä¸€èˆ¬æ高了三å€å·¦å»I¼š(x¨¬)
| Name | SMILES | Correct | FP | Triage | Before | After | Latest |
| Propane | CCC | 65337 | 66352 | 42411 | 42.59 | 17.99 | 14.34 |
| Selenium | [Se] | 246 | 995 | 225 | 0.80 | 0.83 | 0.52 |
| Benzene | c1ccccc1 | 79426 | 79486 | 50893 | 72.69 | 27.56 | 20.29 |
| Methane | C | 118519 | 118524 | 118511 | 61.29 | 5.47 | 4.25 |
| Amido | NC=O | 25695 | 26975 | 14702 | 18.89 | 9.84 | 8.16 |
| Methylbenzene | Cc1ccccc1 | 54529 | 56869 | 20490 | 54.76 | 35.58 | 25.90 |
| Carboxy | OC=O | 33009 | 34369 | 17809 | 23.86 | 12.48 | 10.24 |
| Chlorine | Cl | 19424 | 23318 | 19424 | 11.23 | 1.38 | 1.12 |
| Cyclopropane | C1CC1 | 863 | 4358 | 484 | 8.24 | 7.78 | 5.02 |
| Biphenyl | c1ccccc1c2ccccc2 | 2967 | 5142 | 146 | 21.94 | 21.65 | 11.44 |
| Dopamine | NCCc1ccc(O)c(O)c1 | 829 | 913 | 23 | 1.85 | 2.09 | 1.47 |
| Sulfisoxazole | 7 | 8 | 3 | 0.50 | 0.88 | 0.51 | |
| BetaCarotene | 2 | 16 | 1 | 0.48 | 0.68 | 0.58 | |
| Nitrofurantoin | 0 | 0 | 0 | 0.42 | 0.58 | 0.52 |
A minimal molecular viewer
Click and drag to rotate (left mouse button), zoom/twist (right button) or translate (middle button). The scroll wheel can also be used to zoom.
Developed by Noel O'Boyle (Github repository here). To include a twirlymol in your own web page or blog post, please see this blog post or the HTML and Javascript source of this webpage.
Frowns is loosely based on the PyDaylight API that Andrew Dalke wrote to wrap the daylight C API. In some cases programs written using PyDaylight will also work under Frowns with a few minor changes. A good overview of PyDaylight is available at here.
A good place to look at what Smarts and Smiles are is at the daylight web site located at http://www.daylight.com/
- Smiles parser
- Smarts substructure searching
- SD file parser with SD field manipulations
- Depiction for SD files with coordinates
- Molecule Fingerprint generation
- Several forms of Ring Detection available
- Simple aromaticity perception
- Everything's a graph (i.e. can form canonical strings from incomplete pieces of a molecule)
- Full source code
- Really bad depiction of arbitray molecules! (requires AT&T's GraphViz)
ã€€ã€€Β β beta beta è´å¡”
ã€€ã€€Γ γ gamma gamma 伽马
ã€€ã€€Δ δ deta delta 徯‚€(g¨¨)›_¡”
ã€€ã€€Ε ε epsilon epsilon 艾普襉Kš†
ã€€ã€€Ζ ζ zeta zeta 截塔
ã€€ã€€Η η eta eta 艑֡”
ã€€ã€€Θ θ theta θita 西塔
ã€€ã€€Ι ι iota iota ¾U¦å¡”
ã€€ã€€Κ κ kappa kappa å¡å¸•
  âˆ?nbsp; λ lambda lambda 兰姆è¾?/p>
ã€€ã€€Μ μ mu miu ¾~?/p>
ã€€ã€€Ν ν nu niu ¾U?/p>
ã€€ã€€Ξ ξ xi ksi å¯å¡ž
ã€€ã€€Ο ο omicron omikron 奥密å¯æˆŽ
  ∏ π pi pai ‹z?/p>
ã€€ã€€Ρ ρ rho rou æŸ?/p>
  âˆ?nbsp; σ sigma sigma è¥¿æ ¼é©?/p>
ã€€ã€€Τ τ tau tau å¥?/p>
ã€€ã€€Υ υ upsilon jupsilon 衣普襉Kš†
ã€€ã€€Φ φ phi fai æ–?/p>
ã€€ã€€Χ χ chi khai å–?/p>
ã€€ã€€Ψ ψ psi psai 普西
ã€€ã€€Ω ω omega omiga ‹Æ§ç±³ä¼?br />
åQ?åQ‰æ•°é‡ç¬¦åøP¼š(x¨¬)如:(x¨¬)iåQ?+iåQŒaåQŒxåQŒè‡ªç„¶å¯¹æ•°åº•eåQŒåœ†å‘¨çŽ‡πã€?br />
åQ?åQ‰è¿½Ž—符åøP¼š(x¨¬)å¦‚åŠ åøP¼ˆåQ‹ï¼‰åQŒå‡åøP¼ˆåQ)åQŒä¹˜åøP¼ˆ×æˆ?#183;åQ‰ï¼Œé™¤å·åQ?#247;或ï¼åQ‰ï¼Œä¸¤ä¸ªé›†åˆçš„åƈ集(∪åQ‰ï¼Œäº¤é›†åQ?#8745;åQ‰ï¼Œæ ¹å·åQ?#8730;åQ‰ï¼Œå¯ÒŽ(gu¨©)•°åQˆlogåQŒlgåQŒlnåQ‰ï¼Œæ¯”(åQšï¼‰åQŒå¾®åˆ†ï¼ˆdxåQ‰ï¼Œ¿U¯åˆ†åQ?#8747;åQ‰ç‰ã€?br />
åQ?åQ‰å…³¾pȬ¦åøP¼š(x¨¬)å¦?#8220;åQ?#8221;是ç‰åøP¼Œ“â‰?#8221;是近似符åøP¼Œ“≠”是丽{‰å·åQ?#8220;åQ?#8221;是大于符åøP¼Œ“åQ?#8221;是å°äºŽç¬¦åøP¼Œ“→ ”表示å˜é‡å˜åŒ–的趋势,“âˆ?#8221;是相似符åøP¼Œ“â‰?#8221;是全½{‰å·åQ?#8220;âˆ?#8221;是åã^行符åøP¼Œ“⊥”是垂直符åøP¼Œ“∝”是å比例½W¦å·åQ?#8220;∈”是属于符åøP¼Œ“C”æˆ?#8220;C下é¢åŠ 一æ¨?#8221;æ˜?#8220;包嫔½W¦å·½{‰ã€?br />
åQ?åQ‰ç»“åˆç¬¦åøP¼š(x¨¬)如圆括巓åQˆï¼‰”æ–ÒŽ(gu¨©)‹¬å?#8220;åQ»ï¼½”åQŒèŠ±æ‹¬å·“ï½?j¨©ng)ï½?#8221;括线“â€?#8221;
åQ?åQ‰æ€§è´¨½W¦å·åQšå¦‚æ£å·“åQ?#8221;åQŒè´Ÿå?#8220;åQ?#8221;åQŒç»å¯¹å€¼ç¬¦å?#8220;â€?#8221;
åQ?åQ‰çœç•¥ç¬¦åøP¼š(x¨¬)如三角åÅžåQˆâ–³åQ‰ï¼Œæ£åëuåQˆsinåQ‰ï¼Œä½™åëuåQˆcosåQ‰ï¼Œx的函敎ͼˆf(x)åQ‰ï¼Œæžé™åQˆlimåQ‰ï¼Œå› äØ“(f¨´)åQˆâˆµåQ‰ï¼Œæ‰€ä»¥ï¼ˆ∴åQ‰ï¼Œæ€Õd’ŒåQˆâˆ‘åQ‰ï¼Œ˜qžä¹˜åQ?#8719;åQ‰ï¼Œä»Žnä¸ªå…ƒç´ ä¸æ¯æ¬¡å–出rä¸ªå…ƒç´ æ‰€æœ‰ä¸åŒçš„¾l„åˆæ•ŽÍ¼ˆC(r)(n) åQ‰ï¼Œòq‚(AåQŒAcåQŒAqåQŒx^nåQ‰ï¼Œé˜¶ä¹˜åQˆï¼åQ‰ç‰ã€?br />
数妽W¦å·çš„æ„ä¹?/strong>
½W¦å·ã€€æ„义
∞ã€€æ— ç©·å¤?br />
π  圆周çŽ?br />
|x| ¾l对å€?br />
∪ òq‰™›†
∩ 交集
≥ 大于½{‰äºŽ
≤ ž®äºŽ½{‰äºŽ
≡ æ’ç‰äºŽæˆ–åŒä½™
ln(x) 以e为底的对�
lg(x) �0为底的对�br />
floor(x) 上å–整函æ•?br />
ceil(x) 下å–整函æ•?br />
x mod y 求余�br />
x - floor(x) ž®æ•°éƒ¨åˆ†
∫f(x)dx ä¸å®š¿U¯åˆ†
∫[a:b]f(x)dx a到b的定¿U¯åˆ†
数妽W¦å·çš„应ç”?/strong>
P为真½{‰äºŽ1å¦åˆ™½{‰äºŽ0
∑[1≤k≤n]f(k) 对n˜q›è¡Œæ±‚å’Œ,å¯ä»¥æ‹“广臛_¾ˆå¤šæƒ…å†?br />
如:(x¨¬)∑[n is prime][n < 10]f(n)
∑∑[1≤i≤j≤n]n^2
lim f(x) (x->?) 求æžé™?br />
f(z) f关于z的m阶导函数
C(n:m) ¾l„åˆæ•?nä¸å–m
P(n:m) 排列�
m|n m整除n
m⊥n m与n互质
a ∈ A a属于集åˆA
#A 集åˆAä¸çš„å…ƒç´ ä¸ªæ•°
æ£å¦‚在它自己的网站上比喻的那æ øP¼ŒPallasž®±åƒæ˜¯æ?zh¨¨n)¬å´–å³å£ä¸Šçš„裂¾~,很窄åQŒï¼ˆæŒ‡å®ƒçš„应用领域åƈä¸å¹¿æ³›ï¼‰åQŒä½†å¯¹äºŽæƒ›_ˆ°è¾‘Ö±±™å¶çš„人æ¥è¯ß_(d¨¢)¼ŒåQˆæŒ‡æœ€¾lˆæˆåŠŸè®¾è®¡è¯ç‰©çš„人)åQŒå´æ˜¯å¿…ä¸å¯ž®‘çš„ã€?br />
Pallas主è¦æ˜¯ç”¨äºŽADMET的预‹¹‹ã€‚其包å«çš„å„个模å—相对独立åˆå¯†åˆ‡è”ç³»ã€?br />
包括影å“ADME性质的基¼‹€å¸¸æ•°çš„预‹¹‹ï¼š(x¨¬)pKalcã€PrologPã€PrologDã€Rule of 5ã€TPSAåQ›ä»£è°¢é¢„‹¹‹ï¼š(x¨¬)MetabolExpertã€MEXAlertã€RetroMEXåQ›æ¯’性预‹¹‹ï¼š(x¨¬)HazardExpertã€ToxAlertã€Cytoxicityã€?img src ="http://www.aygfsteel.com/rain1102/aggbug/347986.html" width = "1" height = "1" />
计算æ’äšgåQˆè„‚水分é…ç³»æ•?/span>/考虑ç”?sh¨´)解时的脂水分酾pÀL•°ã€æžæ€§è¡¨é¢ç§¯ã€æº¶è§£æ€§ã€ç”µ(sh¨´)解常数ã€?/span>Lipinski五规则)
All are supported except solubility, in JChemBase, Cartridge, Knime, Pipeline pilot, Instant JChem, Jchem for Excel and in Marvin. See full list of our property predictors.Calculating Lipinski rule of 5:
2. Bulid and maintain project data viewer (SAR understanding)
SAR: structure-activity relationship, ¾l“果与活性关¾p»ï¼Œ½Ž€¿U°æž„效关¾p?/span>
We have R-group decomposition, also a viewer in JChem for Excel, LibMCS GUI. That can be used for SAR understanding.
3. Library enumeration, cleanup, profile and analysis
Reactor, Screen, Calculator plugins, Markush Enumeration, Instant JChem, JChem for Excel, KNIME, Pipeline pilot
Some presentations on the topic:
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
Library Compound Design Methods for CustomLibrary Synthesis
4. Customized Spotfire view
Yes this is the TIBCO Spotfire tool. Marvin is integrated into Spotfire, I think even JChem Cartridge can communicate with Spotfire, our new project is Instant JChem Integration which is under development
5.Similarity search
Yes, JChemBase, Cartridge, Instant JChem, JChem for Excel Similarity search in databases
For a more sophisticated approach of similarity, we provide the Screen package.
6.Clustering
JKlustor, LibMCS
7.Generate SAR Tables
生æˆæž„æ•ˆå…³ç³»è¡¨æ ¼
We do not support directly but we have Rgroup decomposition, Fragmentation toolkit that can be visualized and analysed later.
8.Ligand binding Efficiency
é…体¾l“åˆæ•ˆæžœ
LE can be calculated if the database contains the activity value, heavy atom counts can be calculated in JChem for Excel, Instant Jchem
9.Structure visualization
¾l“æž„å¯è§†åŒ?/span>
Marvin
10.Overlay/Docking
å åˆ/å¯ÒŽ(gu¨©)Ž¥
No, we do not support docking. Alignment can be done in Marvin, Screen3D, and a standalone GUI for low throughput screening.
11.Build predictive ADMET models
建立预测ADMET模型ã€?/span>ADMET分别代表å¸æ”¶ã€åˆ†å¸ƒã€ä»£è°¢ã€æŽ’泄和毒性ã€?/span>
We do not support directly, although we have some calculation plugins that can be further used for these property calculations such as pKa, logP/D, Atom counts, PSA.
EC50åQšåŠæ•°æ•ˆåº”浓度,引è“vå—试对象50%个体产生一¿U特定效应的è¯ç‰©å‰‚é‡ã€?br />
在动物急性毒性试验ä¸åQŒä‹Éå—试动物åŠæ•°æÖMº¡çš„毒物浓度,ç”?LC50表示ã€?br /> åŠæ•°è‡´æ»‹¹“度是衡é‡å˜åœ¨äºŽæ°´ä¸çš„毒物对水生动物和å˜åœ¨äºŽ½Iºæ°”ä¸çš„毒物对哺乛_Š¨ç‰©ä¹ƒè‡³äh¾cÈš„毒性大ž®çš„é‡è¦å‚数。毒物的致æ»æ•ˆåº”与å—试动物暴露时间有密切关系。如果用 LC50表示水ä¸æ¯’物å¯ÒŽ(gu¨©)°´ç”Ÿç”Ÿç‰©çš„急性毒性,必须在LC50å‰æ ‡æ˜Žæš´éœ²æ—¶é—ß_(d¨¢)¼Œå¦?4ž®æ—¶LC50ã€?8ž®æ—¶LC50å’?6ž®æ—¶LC50½{‰ã€‚如果用LC50表示½Iºæ°”ä¸æ¯’物对å“ÞZã^动物的急性毒性,一般是指å—试动物å¸å…¥æ¯’ç‰?ž®æ—¶æˆ?ž®æ—¶åŽçš„试验¾l“æžœåQ?br />
IC50是指“å应”被抑制一åŠæ—¶æŠ‘制剂的‹¹“度åQŒè¿™é‡Œçš„å应å¯ä»¥æ˜¯é…¶å‚¬åŒ–å应åQŒæŠ—原抗体å应ç‰ã€‚在凋亡斚w¢åQŒå¯ä»¥ç†è§£äØ“(f¨´)一定浓度的æŸç§è¯ç‰©è¯±å¯¼è‚¿ç˜¤¾l†èƒž(y¨u)凋亡50%åQŒè¯¥‹¹“度¿UîCØ“(f¨´)50%抑制‹¹“度åQŒå³å‡‹äº¡¾l†èƒž(y¨u)与全部细èƒ?y¨u)数之比½{‰äºŽ50%时所对应的浓度,IC50值å¯ä»¥ç”¨æ¥è¡¡é‡è¯ç‰©è¯±å¯¼å‡‹äº¡çš„能力åQŒå³è¯±å¯¼èƒ½åŠ›‘Šå¼ºåQŒè¯¥æ•°å€ÆD¶Šä½?当然也å¯ä»¥åå‘说明柿U细èƒ?y¨u)对è¯ç‰©çš„è€å—½E‹åº¦ã€?br />
EC50是è¯ç‰©å®‰å…¨æ€§æŒ‡æ ‡ã€‚å…¶å«ä¹‰æ˜¯ï¼š(x¨¬)引è“v50%个体有效的剂é‡æ‰©å¤§å¤šž®‘å€ï¼Œæ‰ä¼š(x¨¬)å¯ÆD‡´50%的个体æ»äº¡ï¼ˆæˆ–ä¸æ¯’)。æ¤å€ÆD¶Šå¤§ï¼Œç”¨è¯ç›¸å¯¹‘Šå®‰å…¨ã€?br /> LD50/ED50ã€TD50/ED50ã€TC50/EC50½{‰ç»Ÿ¿UîCØ“(f¨´)æ²È–—指数åQŒæ˜¯ä¸€¾c»è¯ç‰©çš„å®‰å…¨æŒ‡æ ‡åQŒé€šå¸¸å…¶å€ÆD¶Šå¤§è¶Šå®‰å…¨ã€‚需注愘q™äº›æŒ‡æ ‡åªåæ˜ æ²»ç–—ä½œç”¨ä¸Žæ€¥æ€§æ¯’æ€§çš„å…³ç³»åQŒåƈä¸èƒ½åæ˜ æ…¢æ€§æ¯’æ€§ã€è¿‡æ•æ€§ã€?img src ="http://www.aygfsteel.com/rain1102/aggbug/325622.html" width = "1" height = "1" />
˜q™æ˜¯bioradå…¬å¸ä¸ÞZº†æŽ¨å‡ºæ–îCñ”å“而编写的一首PCR之æŒåQŒä¸€¾Ÿ¤å¯çˆÞqš„人把PCR的过½E‹å”±çš„如æ¤æ·±æƒ…,好åƒæ˜¯P疯的感觉ã€?br />
英文æŒè¯åQ?/strong>
There was a time when to amplify DNA,
You had to grow tons and tons of tiny cells.
Then along came a guy named Dr. Kary Mullis,
Said you can amplify in vitro just as well.
Just mix your template with a buffer and some primers,
Nucleotides and polymerases, too.
Denaturing, annealing, and extending.
Well it’s amazing what heating and cooling and heating will do.
PCR, when you need to detect mutations.
PCR, when you need to recombine.
PCR, when you need to find out who the daddy is.
PCR, when you need to solve a crime
ä¸æ–‡æŒè¯åQ?/strong>
从å‰ä½ è¦æ‰©å¢žDNAåQ?br /> æ€ÀLœ‰åŸ¹å…»å¤§æ‰¹¾l†èƒž(y¨u)çš„é‡ä»»è¦ä½ 背ã€?br /> ˜q™æ—¶æœ‰ä¸ªå®¶ä¼™å«Kary Mullisåšå£«çš„,
他钻出æ¥è¯´ä½“外åˆæˆä¹Ÿä¸€æ ·OKåQ?br /> åªè¦æŠŠä½ 的模æ¿æ؜上缓冲液åQŒå†åŠ 些引物åQ?br /> ˜q˜æœ‰æ ¸è‹·å’Œèšåˆé…¶ã€?br /> å˜æ€§ï¼Œé€€ç«ï¼Œå’Œåšg伸ã€?br /> åŠ çƒ~冷å´~åŠ çƒ~太神奇了åQ?br /> å½“ä½ æƒŒ™¦‹‚€(g¨¨)‹¹‹çªå˜ï¼Œæ¥å•ŠåšPCRå§ï¼
å½“ä½ æƒŒ™¦åŸºå› é‡ç»„åQŒæ¥å•ŠåšPCRå§ï¼
å½“ä½ æƒ³çŸ¥é“å©å他爹到底是è°ï¼Œæ¥å•ŠåšPCRå§ï¼
å½“ä½ æƒŒ™¦ä¾¦ç ´å¤§æ¡ˆåQŒæ¥å•ŠåšPCRå§ï¼
二:(x¨¬)常用QSARæ–ÒŽ(gu¨©)³•ä»‹ç»
1 二维定é‡æž„效关系æ–ÒŽ(gu¨©)³•åQ?D-QSARåQ?br /> ä¼ ç»Ÿçš„äºŒ¾l´å®šé‡æž„效关¾pÀL–¹æ³•å¾ˆå¤šï¼Œæœ‰Hansch法ã€æ¨¡å¼è¯†åˆ«Free-Wilson法和ç”?sh¨´)å拓扑法ã€?å…¶ä¸æœ€ä¸ºé—»å应用最为广泛的ž®±æ˜¯Hanschå’ŒFujitaæ出的Hansch法。它å‡è®¾åŒç³»åˆ—化åˆç‰©æŸäº›ç”Ÿç‰©‹zÀL€§çš„å˜åŒ–是和它们æŸäº›å¯æµ‹é‡ç‰©ç†åŒ–å¦æ€§è´¨çš„å˜åŒ–相è”系的。这些寋¹‹é‡çš„特性包括ç–水性ã€ç”µ(sh¨´)性质和空间立体性质½{‰ï¼Œéƒ½æœ‰å¯èƒ½å½±å“化åˆç‰©çš„生物‹zÀL€§ã€‚Hansch法å‡å®šè¿™äº›å› å是彼æ¤å¤ç«‹çš„,故采用多é‡è‡ªç”Þpƒ½ç›¸å…³æ³•ï¼Œå€ŸåŠ©å¤šé‡¾U¿æ€§å›žå½’牾lŸè®¡æ–ÒŽ(gu¨©)³•ž®±å¯ä»¥å¾—到定é‡æž„效关¾pÀL¨¡åž‹ã€‚Hansch法最åˆå¯ä»¥è¡¨è¾¾äØ“(f¨´)下é¢çš„å…¬å¼?br /> lg1/c=algp+bσ+cE+……+constant
既活性和ç–水性å‚æ•?#960;或lgpã€ç”µ(sh¨´)负性å‚æ•?#963;以åŠ(qi¨¢ng)立体å‚æ•°E有关。åŽæ¥Hanschå‘现è¯ç‰©è¦äº¤æ›¿ç©¿˜q‡æ°´ç›¸å’Œ¾c»è„‚æž„æˆçš„体¾p»ï¼Œå…¶ç§»åŠ¨éš¾æ˜“程度和lgp呈现出函数关¾p…R€‚å‡å¦‚绘q‡ä¸€å®šæ—¶é—´åŽè¯ç‰©åœ¨æœ€æœ«ä¸€ç›æ€¸ä¸ºæµ“度lgcåQŒåˆ™ä»¥lgc对lgp作图åQŒå¯ä»¥å‘现它们之间呈抛物¾U¿å…³¾p»ï¼Œå› æ¤ä¸Šå¼åˆå¯ä»¥å†™æˆä¸‹é¢çš„å½¢å¼
lg1/c=aåQˆlgpåQ?+b lgp +cσ+cE+……+constant ½W¬ä¸€ä¸ªå¼å适用于体外活性数æ®ï¼Œè€Œç¬¬äºŒä¸ªå¼å适用于体内活性数æ®ã€?br /> Hanschå’ŒFujita½{‰äh最åˆæ‰€é‡‡ç”¨çš„构效关¾pÀL¨¡åž‹ä¸åQŒä»…采用了一些简å•çš„分åå‚数。但对于一个分åæ¥è¯ß_(d¨¢)¼Œå¯ä»¥ç”¨å¾ˆå¤šåˆ†åå‚æ•°æ¥è¡¨ç¤ºåˆ†åçš„ä¸åŒç‰¹å¾ï¼Œæ¯”如å„ç§æ‹“扑å‚æ•°ã€çƒåŠ›å¦å‚æ•°ã€é‡åŒ–计½Ž—得到的å‚数以åŠ(qi¨¢ng)分å外åÅžå‚æ•°½{‰ã€‚ç ”½I¶ç»“果表明用˜q™äº›å‚数往往能得到更好的¾l“æžœã€‚å› æ¤åœ¨å®žé™…应用˜q‡ç¨‹ä¸ï¼Œæˆ‘们æ€ÀL˜¯ž®½é‡é€‰æ‹©æœ€ä½?j¨©ng)_‚æ•°æ¥å¾—到最有效的模型而ä¸å¿…å±€é™äºŽHanschå’ŒFujita所æ出的å‚æ•°ã€?br /> æ¤å¤–é™¤äº†ä¼ ç»Ÿçš„çº¿æ€§å›žå½’æ–¹æ³•ä¹‹å¤–ï¼Œä¸€äº›æ–°çš„æ•°ç†ç»Ÿè®¡æ–¹æ³•éžæ•°å€¼ç®—æ³•ä¹Ÿè¢«ç”¨äºŽæž„æ•ˆç ”½I¶ä¸åQŒå¦‚å最ž®äºŒä¹˜æ³•ã€äh工神¾l网¾lœåŠ(qi¨¢ng)é—ä¼ ½Ž—法½{‰ï¼Œ˜q™äº›æ–°æ–¹æ³•çš„应用大大推动2D- QSARæ–ÒŽ(gu¨©)³•çš„å‘展ã€?br />
2 三维定é‡æž„效关系æ–ÒŽ(gu¨©)³•åQ?D-QSARåQ?br /> ˜q‘些òq´æ¥åQŒéšç€æž„效关系ç†è®ºå’Œç»Ÿè®¡æ–¹æ³•çš„˜q›ä¸€æ¥å‘展,åˆå‡ºçŽîCº†ä¸€äº›ä¸‰¾l´å®šé‡æž„效关¾p»ï¼ˆ3D-QSARåQ‰æ–¹æ³•ã€‚è¬å¦‚分å外形分æžï¼ˆmolecularshape analysis, MSAåQ‰ã€è·¼›Õd‡ 何方æ³?distance geometry, DG)和以å?qi¨¢ng)比较分å场分æžåQˆcomparative molecular field analysis, CoMFAåQ‰æ–¹æ³•ã€?br /> ä¸?D-QSAR比较åQ?D-QSARæ–ÒŽ(gu¨©)³•åœ¨ç‰©ç†åŒ–å¦ä¸Šçš„æ„义更为明¼‹®ï¼Œèƒ½é—´æŽ¥åæ˜ è¯ç‰©åˆ†åå’Œé¶ç‚¹ä¹‹é—´çš„éžé”®ç›¸äº’作用特å¾ã€‚å› æ¤è¿‘å多òq´æ¥3D-QSARæ–ÒŽ(gu¨©)³•å¾—到了迅速的å‘展和广泛的应用。在3D-QSARæ–ÒŽ(gu¨©)³•ä¸ï¼Œæ¯”较分å场分æžï¼ˆcomparative molecular field analysis, CoMFAåQ‰æ–¹æ³•æ˜¯ç›®å‰æœ€ä¸ºæˆç†Ÿä¸”应用最为广泛的æ–ÒŽ(gu¨©)³•ã€?br />
2.1 比较分å场分æžï¼ˆcomparative molecular field analysis, CoMFAåQ‰æ–¹æ³•ç®€ä»?br /> CoMFA的基本原ç†æ˜¯åQšå‡å¦‚一¾l„相似化åˆç‰©ä»¥åŒæ ïL(f¨¥ng)š„æ–¹å¼ä½œç”¨äºŽåŒä¸€é¶ç‚¹åQŒé‚£ä¹ˆå®ƒä»¬çš„生物‹zÀL€§å°±å–决于æ¯ä¸ªåŒ–åˆç‰©å››å‘¨åˆ†å场的差别åQŒè¿™¿U分å场å¯ä»¥åæ˜ è¯ç‰©åˆ†åå’Œé¶ç‚¹ä¹‹é—´æŒ‰çš„éžé”®ç›¸äº’作用特性。其计算å¯ä»¥½Ž€å•çš„分äØ“(f¨´)三个æ¥éª¤åQ?åQ‰é¦–先确定è¯ç‰©åˆ†åçš„‹zÀL€§æž„象,å†æŒ‰ä¸€å®šçš„规则åQˆä¸€èˆ¬äØ“(f¨´)骨架å åŠ æˆ–åœºå åŠ åQ‰è¿›è¡Œè¯ç‰©åˆ†åçš„å åˆåQ›ï¼ˆ2åQ‰ç„¶åŽï¼Œåœ¨å åˆå¥½çš„分å四周定义一定的æ¥é•¿å‡åŒ€åˆ’åˆ†äº§ç”Ÿæ ¼ç‚¹åQŒåœ¨æ¯ä¸ªæ ¼ç‚¹ä¸Šç”¨ä¸€ä¸ªæŽ¢é’ˆç¦»åæ¥è¯„äh(hu¨¢n)æ ¼ç‚¹ä¸Šçš„åˆ†å场特å¾ï¼ˆä¸€èˆ¬äØ“(f¨´)é™ç”µ(sh¨´)场和立体场,有时也包括ç–水场和氢键场åQ‰ï¼›åQ?åQ‰æœ€åŽé€šè¿‡å最ž®äºŒä¹˜æ–¹æ³•å¾ç«‹åŒ–åˆç‰©‹zÀL€§å’Œåˆ†å场特å¾ä¹‹é—´çš„关系òq¶ç»™å‡ºå„¿U分åé¢çš„ç‰åŠ¿èƒ½é¢ã€?br /> ˜q‘å¹´æ¥ï¼Œç ”ç©¶äººå‘˜å¯¹ä¼ ¾lŸçš„CoMFA˜q›è¡Œäº†å¤§é‡çš„改进åQŒå…¶ä¸æ¶‰å?qi¨¢ng)到‹zÀL€§æž„象的¼‹®å®šåQŒåˆ†åå åŠ è§„åˆ™ã€åˆ†å场势函数的定义以åŠ(qi¨¢ng)分å场å˜é‡çš„选喽{‰ç‰åQŒåœ¨å¾ˆå¤§½E‹åº¦ä¸Šæ高了CoMFA计算的æˆåŠŸçŽ‡ã€‚å…¶ä¸æœ€å…ähœ‰ä»£è¡¨æ€§çš„å¯èƒ½ž®±æ˜¯æ¯”较分åç›æ€¼¼å› å分æžåQˆcomparative molecular similarity indices analysis, CoMSIAåQ‰æ–¹æ³•ã€?br />
2.2 比较分åç›æ€¼¼å› å分æžåQˆcomparative molecular similarity indices analysis, CoMSIAåQ‰æ–¹æ³•ç®€ä»?br /> 与CoMFAæ–ÒŽ(gu¨©)³•ç›¸æ¯”åQŒæœ€å¤§çš„ä¸åŒž®±æ˜¯åˆ†å场的能é‡å‡½æ•°é‡‡ç”¨äº†ä¸Žè·ç¦»ç›¸å…³çš„高斯函数的形å¼åQŒè€Œä¸æ˜¯ä¼ ¾lŸçš„Coulomb å’ŒLennard-Jones 6-12势函数的形å¼ã€‚CoMSIAæ–ÒŽ(gu¨©)³•ä¸å…±å®šä¹‰äº”ç§åˆ†å场的特å¾åQŒåŒ…括立体场ã€é™ç”?sh¨´)场ã€ç–水场以åŠ(qi¨¢ng)氢键场(包括氢键¾l™ä½“场和氢键å—体场)。这五ç§åˆ†å场å¯ä»¥é€šè¿‡å…¬å¼è®¡ç®—得到。在CoMSIAæ–ÒŽ(gu¨©)³•ä¸ï¼Œç”׃ºŽé‡‡ç”¨äº†ä¸Žè·ç¦»ç›¸å…³çš„高斯函数åÅžå¼ï¼Œå¯ä»¥æœ‰æ•ˆåœ°é¿å…åœ¨ä¼ ç»ŸCoMFAæ–ÒŽ(gu¨©)³•ä¸ç”±é™ç”µ(sh¨´)场和立体场的函数形å¼æ‰€å¼•è“v的缺陗÷€‚由于分å场能é‡åœ¨æ ¼ç‚¹ä¸Šçš„迅速衰退åQŒä¸éœ€è¦å®šä¹‰èƒ½é‡çš„截æ–åQˆcutoffåQ‰å€? 对一些实际体¾p»è¿›è¡Œäº†˜q™ä¸¤¿U方法的比较¾l“果分æžåQŒåœ¨è®¡ç®—ä¸é‡‡ç”¨äº†ä¸åŒçš„æ ¼ç‚ÒŽ(gu¨©)•°åQŒä¸”对体¾pÕd‡é‡‡ç”¨å…¨ç©ºé—´æœç´¢ç–略。结果表明CoMFA计算对ä¸åŒçš„æ ¼ç‚¹å¤§å°å€ég»¥å?qi¨¢ng)å åˆåˆ†åä¸åŒçš„½Iºé—´å–å‘éžå¸¸æ•æ„ŸåQŒé‡‡ç”¨ä¸åŒçš„½Iºé—´å–å‘æ—Óž¼Œå›žå½’¾pÀL•°çš„差值最大å¯ä»¥è¾¾åˆ?.3以上。而CoMSIAæ–ÒŽ(gu¨©)³•åœ¨è®¡½Ž—ä¸åŒæ ¼ç‚¹å¤§ž®å–å€ég»¥å?qi¨¢ng)分å空间å–å‘下得到的结果则½E›_®šçš„多åQŒåœ¨ä¸€èˆ¬æƒ…况下åQŒCoMSIA计算ä¼?x¨¬)å¾—åˆ°æ›´åŠ æ»¡‘³çš?D-QSAR模型ã€?br />
3. 4D-QSARæ–ÒŽ(gu¨©)³•½Ž€ä»?br /> 1997òqß_(d¨¢)¼Œhopfinger½{‰æå‡ÞZº†4D-QSAR的概å¿üc(di¨£n)€‚作者首‹Æ¡é‡‡ç”¨é—ä¼ ç®—æ³•é€‰æ‹©åˆ†å动力å¦äñ”生的构象æ¥äñ”生最佳的构效关系模型。在˜q™ä¸ªæ–ÒŽ(gu¨©)³•ä¸ï¼Œä½œè€…用æ¯ä¸ªæ ¼ç‚¹å¯¹ç”¨çš„原åå 有率æ¥ä½œä¸ºPLSçš„å˜é‡ï¼Œä½œè€…æ ¹æ®åŽŸåçš„ä¸åŒç‰¹å¾å®šä¹‰äº†ä¸ƒ¿Uä¸åŒç§¾cÈš„原å模型。在4D-QSARæ–ÒŽ(gu¨©)³•ä¸ï¼Œä½œè€…考虑了è¯ç‰©åˆ†å的整个构象½Iºé—´åQŒè€Œä¸æ˜¯ä¸€ä¸ªåˆ†å,而且考察了多¿U原åå åˆæ–¹å¼ï¼Œå› æ¤åœ¨æ¦‚å¿µä¸Šæ¯”ä¼ ¾lŸçš„CoMFAæ–ÒŽ(gu¨©)³•æœ‰ä¸€å®šçš„˜q›æ¥
ä¸?建立定é‡æž„效关系的方æ³?br />
3.1 ¾U¿æ€§å›žå½’æ–¹æ³?br /> åœ¨ä¼ ¾lŸäºŒ¾l´æž„æ•ˆç ”½I¶ä¸åQŒå¤šé‡çº¿æ€§å›žå½’(multiple linear regression, MLRåQ‰æ–¹æ³•æ˜¯æœ€ä¸ºå¸¸è§çš„¾lŸè®¡æ–ÒŽ(gu¨©)³•ã€‚一个分åå¯ä»¥ç”¨å¾ˆå¤šåˆ†åå‚æ•°æ¥è¡¨è¾¾ï¼Œä½†åœ¨å»ºç«‹å¤šé‡¾U¿æ€§å›žå½’模型的时候,为é¿å…过拟åˆåQˆoverfittingåQ‰ï¼Œæˆ‘们åªèƒ½ä»Žè¿™äº›ç‰©ç†åŒ–å¦å‚æ•îC¸é€‰æ‹©ä¸€éƒ¨åˆ†å‚æ•°æ¥å¾ç«‹å›žå½’模型。一般æ¥è®ÔŒ¼ŒåŒç³»ç‰©æ•°ç›®å’Œæ‰€é€‰å–å‚数数目的比应大äº?ï½?åQŒä¹Ÿæœ‰ähæ出应大äº?çš„n‹Æ¡æ–¹åQˆn表示选å–çš„å‚æ•îC¸ªæ•ŽÍ¼‰åQŒæ€Žæ ·é€‰å–åˆé€‚çš„å‚数一直是定é‡æž„æ•ˆå…³ç³»ç ”ç©¶ä¸çš„一个难题,而且对于¾U¿æ€§å›žå½’æ¥è¯ß_(d¨¢)¼Œå½“体¾pÕd™ªå£°è¾ƒå¼ºæˆ–òq²æ‰°ä¸¥é‡æ—Óž¼Œæœ‰å¯èƒ½å¯¼è‡´æ‰€å¾—的模型å¤ÞqœŸåQŒäØ“(f¨´)了克æœå¤šé‡çº¿æ€§å›žå½’çš„ä¸èƒöåQŒåœ¨æ•°å¦ä¸Šå¯é‡‡ç”¨ä¸ÀLˆåˆ†å›žå½’方法ã€?br /> 所谓主æˆåˆ†å›žå½’ž®±æ˜¯é‡‡ç”¨ä¸ÀLˆåˆ†åˆ†æžæ–¹æ³?principle component analysis, PCA)å¯ÒŽ(gu¨©)´»æ€§åª„å“æœ€å¤§çš„å‡ ä¸ªä¸»è¦æˆåˆ†å»ºç«‹å®šé‡æž„效关系模型。所谓主æˆåˆ†æ˜¯ä¸€¾l„æ–°çš„å˜é‡ï¼Œå®ƒæ˜¯åŽŸæ¥å˜é‡Xij的线性组åˆï¼Œ½W¬ä¸€ä¸ªä¸»æˆåˆ†æ‰€èƒ½è§£é‡ŠåŽŸé‡çš„方差最大,½W¬äºŒä¸ªæ¬¡ä¹‹ï¼Œ½W¬ä¸‰ä¸ªå†‹Æ¡ä¹‹åQŒå¾€ä¸‹ä¾æ¤ç±»æŽ¨ã€‚也ž®±æ˜¯è¯ß_(d¨¢)¼Œä¸ÀLˆåˆ†æ˜¯ä¸€¿U线性组åˆï¼Œç”¨å®ƒæ¥è¡¨½CºåŽŸæ¥å˜é‡æ‰€äº§ç”Ÿçš„åã^方误差最ž®ã€‚è¿ç”¨ä¸»æˆåˆ†åˆ†æžåQŒåŽŸå˜é‡çŸ©é˜µXå¯ä»¥è¡¨è¾¾ä¸ºå¾—分(å³ä¸»æˆåˆ†åQ‰çŸ©é˜µT, 该矩é˜ëŠ”±æœ¬å¾çŸ¢é‡ä¸Šçš„投媄所得。主矩阵与矩é˜ëŠš„本å¾çŸ¢é‡ä¸€ä¸€å¯¹åº”åQŒå³XP=T。主æˆåˆ†å›žå½’å¯ä»¥æœ‰æ•ˆè§£å†³å…Þqº¿é—®é¢˜åQŒåŒæ—¶ç”±äºŽåŽ»æŽ‰äº†ä¸å¤ªé‡è¦çš„主æˆåˆ†åQŒå› 而å¯ä»¥å‰Šå¼±å™ªå£ŽÍ¼ˆéšæœºè¯¯å·®åQ‰æ‰€äº§ç”Ÿçš„媄å“。主æˆåˆ†å›žå½’å¯ä»¥åˆ†äØ“(f¨´)两æ¥åQ? ‹¹‹å®šä¸ÀLˆåˆ†æ•°åQŒåƈ通过ä¸ÀLˆåˆ†åˆ†æžå°†X矩阵é™ç»´åQ? 对于é™ç»´çš„X矩阵å†åš¾U¿æ€§å›žå½’分æžã€‚但是å‡å¦‚在½W¬ä¸€æ¥æ¶ˆåŽÈš„是有用的ä¸ÀLˆåˆ†ï¼Œè€Œä¿ç•™çš„是噪壎ͼŒåˆ™ç¬¬äºŒæ¥å¤šå…ƒ¾U¿æ€§å›žå½’所得的¾l“æžœž®±å°†å离真实的数å¦æ¨¡åž‹ã€?br /> 在主æˆåˆ†å›žå½’法的½W¬ä¸€æ¥ä¸åQŒæˆ‘们处ç†çš„仅是X矩阵åQŒå¯¹äºŽçŸ©é˜µYä¸çš„ä¿¡æ¯òq¶æœªè€ƒè™‘。事实上åQŒYä¸å¯èƒ½åŒ…å«æœ‰ç”¨çš„ä¿¡æ¯åQŒæ‰€ä»¥ä¸€¿U很自然的想法是‹¹‹è¯•çŸ©é˜µXå› åæ—¶åŒæ—¶è€ƒè™‘矩阵Y的作用。å最ž®äºŒä¹˜æ³•åœ¨è€ƒè™‘自å˜é‡çš„åŒæ—¶ä¹Ÿè€ƒè™‘å› å˜é‡çš„作用åQŒåŒæ—‰™€šè¿‡æŠ˜è¡·å„自½Iºé—´å†…çš„å› å使模型较好的åŒæ—¶æ述自å˜é‡å’Œå› å˜é‡ï¼Œä»Žè€Œæœ‰æ•ˆåœ°å‡å°‘ç›¸å…³å› ç´ çš„åª„å“ã€?br /> 在构效关¾pÈ ”½I¶è¿‡½E‹ä¸åQŒè¿˜ç”¨åˆ°äº†è®¸å¤šå…¶ä»–的数熾lŸè®¡æ–ÒŽ(gu¨©)³•åQŒæ¯”如说é€æ¥å›žå½’æ–ÒŽ(gu¨©)³•ã€éž¾U¿æ€§æœ€ž®äºŒä¹˜æ–¹æ³•ä»¥å?qi¨¢ng)一些模å¼è¯†åˆ«æ–¹æ³•ç‰åQŒåœ¨æ¤å°±ä¸ä¸€ä¸€ä»‹ç»ã€?br />
3.2 é—ä¼ ½Ž—法
3.2.1é—ä¼ ½Ž—法½Ž€ä»?br /> é—ä¼ ½Ž—法åQˆgenetic algorithm, GAåQ‰åŸºæœ¬æ€æƒ³æºè‡ªäºŽè¾¾ž®?d¨¡ng)文的自焉™€‰æ‹©å¦è¯´ã€‚它表明é—ä¼ å’Œå˜å¼‚是军_®šç”Ÿç‰©˜q›åŒ–çš„å†…åœ¨å› ç´ ï¼Œç”Ÿç‰©çš„é—ä¼ ç‰¹æ€§ï¼Œä½¿ç”Ÿç‰©ç•Œçš„ç‰©¿Uä¿æŒç›¸å¯¹ç¨³å®šï¼Œè€Œå˜å¼‚特性则使生物个体äñ”生新的性状åQŒä»¥è‡³äºŽå½¢æˆæ–°çš„物ç§åQŒæŽ¨åŠ¨äº†ç”Ÿç‰©çš„进化和å‘展ã€?br /> é—ä¼ ½Ž—法是模拟达ž®?d¨¡ng)æ–‡é—ä¼ é€‰æ‹©å’Œè‡ªç„¶æ·˜æ±°ç”Ÿç‰©è¿›åŒ–è¿‡½E‹çš„计算模型。它的æ€æƒ³æºè‡ªäºŽç”Ÿç‰©é—ä¼ å¦å’Œé€‚者生å˜çš„自然规律åQŒæ˜¯å…ähœ‰“生å˜+‹‚€(g¨¨)‹¹?#8221;˜q代˜q‡ç¨‹çš„æœç´¢ç®—法。é—ä¼ ç®—æ³•ä»¥ä¸€¿U群体ä¸çš„所有个体äØ“(f¨´)对象åQŒåƈ利用éšæœºåŒ–技术指导对一个被¾~–ç çš„å‚数空间进行高效æœç´¢ã€‚å…¶ä¸ï¼Œé€‰æ‹©ã€äº¤å‰å’Œå˜å¼‚æž„æˆäº†é—ä¼ ç®—æ³•çš„é—ä¼ æ“作。å‚æ•°ç¼–ç ã€åˆå§‹ç¾¤ä½“的讑֮šã€é€‚应度函数的讑֮šã€é—ä¼ æ“作设计ã€æŽ§åˆ¶å‚数设定五个è¦ç´ 组æˆäº†é—ä¼ ½Ž—æ³•çš„æ ¸å¿ƒå†…å®V€?br /> é—ä¼ ½Ž—法的主è¦ç‰¹ç‚ÒŽ(gu¨©)˜¯1直接对结构对象进行æ“作,ä¸å˜åœ¨æ±‚导和函数˜qžç®‹æ€§çš„é™å®šåQ?å…ähœ‰å†…在的éšòq¶è¡Œæ€§å’Œæ›´å¥½çš„全局å¯ÖM¼˜èƒ½åŠ›åQ? 采用概率化的å¯ÖM¼˜æ–ÒŽ(gu¨©)³•åQŒèƒ½è‡ªåŠ¨èŽ·å–和指å¯ég¼˜åŒ–çš„æœçƒ¦½Iºé—´åQŒè‡ªé€‚应的调整æœç´¢æ–¹å‘,ä¸éœ€è¦ç¡®å®šçš„规则。é—ä¼ ç®—æ³•çš„˜q™äº›æ€§è´¨å·²è¢«äºÞZ»¬òq¿æ³›çš„用于组åˆä¼˜åŒ–ã€æœºå™¨å¦ä¹?f¨¤n)ã€ä¿¡å·å¤„ç†ã€è‡ªé€‚应控制和äh工生命ç‰é¢†åŸŸã€‚作ä¸ÞZ¸€¿U新的全局优化æœèŽ·æ–ÒŽ(gu¨©)³•åQŒé—ä¼ ç®—æ³•ä»¥å…¶ç®€å•é€šç”¨ã€é€‚用于åƈ行处ç†ä»¥å?qi¨¢ng)高效ã€å®žç”¨ç‰æ˜¾è‘—特点在å„个领域得åˆîCº†òq¿æ³›çš„应用,å–得了良好的效果åQŒåƈé€æ¸æˆäØ“(f¨´)最为é‡è¦çš„æ™ø™ƒ½½Ž—法之一ã€?br /> é—ä¼ ½Ž—法的兴èµäh˜¯åœ?0òq´ä»£æœ«è‡³90òq´ä»£åˆï¼Œæœ€åˆåªæ˜¯ç”¨äºŽå¤šè‚½çš„构象分æžåQŒè¿‘些年æ¥ï¼Œé—ä¼ ½Ž—法在工业分æžã€å…‰è°±åˆ†æžã€è›‹ç™½è´¨ä¸‰çñ”¾l“构猜测和数æ®åˆ†æžç‰æ–šw¢ä¹Ÿå–得了òq¿æ³›åº”用ã€?br />
3.2.2 é—ä¼ ½Ž—法基本原ç†
é—ä¼ ½Ž—法的原ç†æ¯”较简å•ï¼Œä¼ 统é—ä¼ ½Ž—法¾l常用一个二˜q›åˆ¶¾U¿æ€§æ•°å—串表示一个个体(individualåQ?若干个体构æˆä¸€ä¸ªç§¾Ÿ¤ï¼ˆpopulationåQ?一般我们用éšæœºçš„办法äñ”生åˆå§‹ç§¾Ÿ¤ã€‚åˆå§‹ç§¾Ÿ¤äñ”生以åŽï¼Œž®Þq”¨å®šä¹‰çš?#8220;适应性函æ•?#8221;åQˆfitness functionåQ‰åŽ»è¯„äh(hu¨¢n)¿U群ä¸çš„æ¯ä¸ªä¸ªä½“。然åŽé€šè¿‡ä¸ªä½“的得分选择部分个体òq‰™€šè¿‡äº¤å‰å’Œå˜å¼‚去¾Jè¡å®ƒä»¬çš„åŽä»£ã€‚当然得分高的被选ä¸çš„å¯èƒ½æ€§ä¼š(x¨¬)大些åQŒåŒæ—‰™‚£äº›æœªé€‰ä¸çš„个体就自然消亡。留下æ¥çš„个体和它们æ–îCñ”生的å代ž®±åÅžæˆäº†æ–°çš„¿U群。绘q‡ä¸€äº›åˆ—交å‰å’Œå˜å¼‚过½E‹åŽåQŒå°±å¯ä»¥å¾—到æ€ÖM½“得分比较高的¿U群åQŒè¿™ž®±æ˜¯æˆ‘们所è¦å¯»æ‰„¡š„å…ähœ‰éžå‡¡æ€§èƒ½çš„æž„åž‹ã€?br /> 我们ä¹?f¨¤n)惯上把Holland 1975òq´æ出的GA¿UîCØ“(f¨´)ä¼ ç»Ÿçš„GA。其主è¦æ¥éª¤å¦‚下åQ?¾~–ç . GA在进行æœç´¢ä¹‹å‰å…ˆž®†è§£½Iºé—´çš„解数æ®è¡¨ç¤ºæˆé—ä¼ ç©ºé—´çš„åŸºå› åž‹ä¸²¾l“æž„æ•°æ®åQŒè¿™äº›ä¸²¾l“æž„æ•°æ®çš„ä¸åŒç»„åˆä¾¿æž„æˆäº†ä¸åŒçš„点;2 产生åˆå§‹¿U群。éšæœÞZñ”生åˆå§‹ä¸²¾l“æž„æ•°æ®ã€‚æ¯ä¸ªä¸²¾l“构数殿UîCØ“(f¨´)一个个体,多个个体构æˆä¸€ä¸ªç§¾Ÿ¤ã€‚GA以这个串¾l“æž„æ•°æ®ä½œäØ“(f¨´)åˆå§‹ç‚¹å¼€å§‹è„P代优化过½E‹ï¼›3 ˜q›è¡Œé€‚应性指评估‹‚€(g¨¨)‹¹‹ã€‚适应性函数表明个体或解的优劣性。ä¸åŒçš„问题åQŒé€‚应性函数的定义方å¼ä¹Ÿä¸åŒï¼›4 选择。选择的目的是ä¸ÞZº†ä»Žå½“å‰ç¾¤ä½“ä¸é€‰å‡ºä¼˜è‰¯çš„个体,使它们有æœÞZ¼š(x¨¬)作äØ“(f¨´)父代ä¸ÞZ¸‹ä¸€ä»£ç¹ŒD–åå™ã€‚选择实现了达ž®?d¨¡ng)文的适者生å˜åŽŸåˆ™ï¼›5 交å‰ã€‚交å‰æ“作是é—ä¼ ½Ž—法ä¸æœ€ä¸»è¦çš„é—ä¼ æ“作。通过交å‰æ“作å¯ä»¥å¾—到æ–îC¸€ä»£ä¸ªä½“,æ–îC¸ªä½“组åˆäº†å…¶çˆ¶è¾ˆä¸ªä½“çš„ç‰ÒŽ(gu¨©)€§ï¼Œäº¤å‰ä½“现了信æ¯äº¤æ¢çš„æ„æ€ï¼›6 å˜å¼‚。å˜å¼‚首先在¾Ÿ¤ä½“ä¸éšæœºé€‰æ‹©ä¸€ä¸ªä¸ªä½“,对于选ä¸çš„个体以一定的概率éšæœºåœ°æ”¹å˜ä¸²¾l“æž„æ•°æ®ä¸çš„æŸä¸ªä¸²çš„倹{€‚åŒç”Ÿç‰©ç•Œä¸€æ øP¼Œå˜å¼‚为新个体的äñ”生æ供机ä¼?x¨¬)ã€?br />
3.2.3 é—ä¼ ½Ž—法基本特点
é—ä¼ ½Ž—法作äØ“(f¨´)一¿Uå¿«æ—÷€ç®€ä¾Ñ€å®¹é”™æ€§å¼ºçš„算法,在儾cÈ»“构对象的优化˜q‡ç¨‹ä¸æ˜¾½Cºå‡ºæ˜Žæ˜¾çš„优åŠÑ€‚ä¸Žä¼ ç»Ÿæœçƒ¦æ–ÒŽ(gu¨©)³•ç›¸æ¯”åQŒé—ä¼ ç®—æ³•å…·æœ‰ä»¥ä¸‹ç‰¹ç‚¹ï¼š(x¨¬)1 æœçƒ¦˜q‡ç¨‹ä¸ç›´æŽ¥ä½œç”¨åœ¨å˜é‡ä¸Šï¼Œè€Œæ˜¯ä½œç”¨åœ¨å‚数集˜q›è¡Œäº†ç¼–ç 的个体上。椾~–ç æ“作使得é—ä¼ ½Ž—法å¯ç›´æŽ¥å¯¹¾l“构对象åQˆé›†åˆã€åºåˆ—ã€çŸ©é˜üc(di¨£n)€æ ‘(w¨¨i)ã€å›¾ã€é“¾å’Œè¡¨åQ‰è¿›è¡Œæ“作;2 æœçƒ¦˜q‡ç¨‹æ˜¯ä»Žä¸€¾l„è„P代到å¦ä¸€¾l„解åQŒé‡‡ç”¨åŒæ—¶å¤„ç†ç¾¤ä½“ä¸å¤šä¸ªä¸ªä½“的方法,é™ä½Žäº†é™·å…¥å±€éƒ¨ä¼˜åŒ–çš„å¯èƒ½æ€§ï¼Œòq¶æ˜“于优化;3 采用概率å˜è¿çš„规则æ¥æŒ‡å¯¼æœçƒ¦æ–¹å‘åQŒè€Œä¸é‡‡ç”¨¼‹®å®šæ€§æœç´¢è§„则;4å¯ÒŽ(gu¨©)œç´¢ç©ºé—´æ²¡æœ‰ä“Q何éžå‡¡çš„è¦æ±‚åQˆå¦‚˜qžé€šæ€§ã€å‡¸æ€§ç‰åQ‰ï¼Œåªåˆ©ç”¨é€‚应性信æ¯ï¼Œä¸éœ€è¦å¯¼æ•°ç‰å…¶ä»–辅助信æ¯åQŒé€‚用范围更广ã€?br />
3.2.4 é—ä¼ ½Ž—法的工作原ç?br /> é—ä¼ ½Ž—法是一个以适应度函敎ͼˆæˆ–ç›®æ ‡å‡½æ•ŽÍ¼‰ä¸ÞZ¾æ®ï¼Œé€šè¿‡å¯¹ç¾¤ä½“ä¸ä¸ªä½“æ–½åŠ é—ä¼ æ“作实现¾Ÿ¤ä½“内个体结构釾l„çš„˜q代优化˜q‡ç¨‹ã€‚它的ç†è®ÞZ¸»è¦æœ‰æ¨¡å¼å®šç†ã€ç§¯æœ¨å—å‡è®¾ã€æœ€ž®æ¬ºéª—问题和éšåƈ行性问题ç‰ã€‚我们主è¦ä»‹¾l模å¼ç†è®ºå’Œæœ€ž®æ¬ºéª—问题ã€?br /> 模å¼ç†è®ºæ˜¯ç ”½I¶è¾ƒòqр媄å“较大的GAç†è®ºã€‚它?y¨u)®†GA的追Ž—过½E‹ç†è§£äØ“(f¨´)模å¼æ“作˜q‡ç¨‹òq¶ä»Žæ¨¡å¼˜q算的角度解释GA的性能特点。GA的计½Ž—过½E‹æ˜¯ä»Žä¸€ä¸ªä¸ªä½“组æˆçš„åˆå§‹¾Ÿ¤ä½“出å‘åQŒåó@环的执行选择ã€äº¤å‰ã€å˜å¼‚过½E‹ã€‚表é¢ä¸Šçœ‹è¿½Ž—过½E‹ç›´æŽ¥ä½œç”¨äºŽ¾Ÿ¤ä½“åQŒä½†å…¶ä¸éšå«äº†æ¨¡å¼é›†åˆä¸çš„æ“作过½E‹ã€‚模å¼å®šç†ä»Žæ¨¡å¼æ“作的角度论è¯äº†åœ¨é€‰æ‹©ã€äº¤æ¢å’Œå˜å¼‚å› å作用下æŸä¸€æ¨¡å¼é‡çš„å˜åŒ–。模å¼å®šç†æ½Cºå‡ºå…ähœ‰ä½Žé˜¶ã€çŸå®šä¹‰çŸ©ä»¥å?qi¨¢ng)åã^å‡é€‚应度高于群体åã^å‡é€‚应度的模å¼åœ¨å代丞®†ä»¥æŒ‡æ•°¾U§å¢žé•¿ï¼Œç”׃ºŽé‡ç»„ã€å˜å¼‚的作用åQŒåƈéžä½Žé˜¶ä¼˜äºŽåã^å‡çš„模å¼éƒ½æ˜¯æœ‰æ•ˆæ¨¡å¼åQŒä½†åœ¨è§„模äØ“(f¨´)N的群体ä¸åQŒæœ‰å¤šä¸ªæ¨¡å¼æ˜¯æœ‰æ•ˆçš„åQŒå³æ¯ä¸€ä»£GA处ç†ä¸ªä½“çš„åŒæ—Óž¼ŒèŽ·å¾—了对多个模å¼çš„åƈ行处ç†ï¼Œòq¶ä¸”æ— é¡»é¢å¤–çš„å˜å‚¨é‡åQŒè¿™ä¸€æ€§è´¨¿UîCØ“(f¨´)éšåƈ行性。也ž®±æ˜¯GA在群体ä¸æœçƒ¦çš„过½E‹å¯çœ‹ä½œæ˜¯éšå«çš„å¯ÒŽ(gu¨©)¨¡å¼çš„æŠ½æ ·˜q‡ç¨‹åQŒç„¶åŽé€šè¿‡é—ä¼ ½Ž—å的作用将çŸçš„低阶模å¼çš„ä¿¡æ¯ç»„åˆè“væ¥ï¼Œå½¢æˆé«˜é˜¶æ¨¡å¼åQŒç›´è‡Ïxœç´¢åˆ°æœ€ä¼˜è§£åQŒè¿™è¯´æ˜ŽåŒä¼ ¾lŸçš„优化½Ž—法相比åQŒGA有很强的处ç†èƒ½åŠ›ã€?br /> ç ”ç©¶‹Æºéª—是äØ“(f¨´)了猜‹¹‹ç»™å®šé—®é¢˜ç”¨GA求解的难易程度,˜q™æ˜¯æˆ‘们所å…Ïx³¨çš„问题。由模å¼ç†è®ºåQŒä¸€ä¸ªé—®é¢˜èƒ½å¦ç”¨é—ä¼ ½Ž—法有效地求解,å–决于问题编ç 是å¦èƒ½æ»¡èƒö¿U¯æœ¨å—å‡è®¾ï¼Œæ»¡èƒö者用é—ä¼ ½Ž—法求解率高åQŒä¸æ»¡èƒö者求解率较低åQŒç”šè‡Ïx‰¾ä¸åˆ°æ»¡èƒö解,˜q™ä¸€çŽ°è±¡¿UîCØ“(f¨´)‹Æºéª—。最ž®æ¬ºéª—问题旨在尽å¯èƒ½ä½‰K—®é¢˜çš„¾~–ç ˜qå¿U¯æœ¨å—å‡è®¾ï¼Œä½‰K«˜é˜¶ç§¯æœ¨å—ä¸èƒ½ç”Ÿæˆæœ€ä¼˜è§£ã€‚但实验¾l“果表明åQŒå¯¹åŒ…嫋ƺ骗的问题,用GA求解时得到最优解和得ä¸åˆ°æœ€ä¼˜è§£çš„情况都有å¯èƒ½å‘生。这说明‹Æºéª—ä¸æ˜¯è¡¡é‡ç”¨GA解决问题难易½E‹åº¦çš„å”¯ä¸€æ ‡å‡†ã€‚ç›®å‰æˆ‘ä»¬è¿˜æ— æ³•åˆ¤å®šä¸€ä¸ªé—®é¢˜åŒ…å«æ¬ºéª—的多少和问题相对于GA难易½E‹åº¦é—´çš„关系ã€?br />
3.2.5 é—ä¼ ½Ž—法的一个计½Ž—实ä¾?br /> é—ä¼ ½Ž—法在程åºä¸Šå®žçŽ°òq¶ä¸å¤æ‚åQŒæˆ‘们å¯ä»¥é€šè¿‡è€ƒè™‘如下½Ž€å•çš„éžçº¿æ€§ä¼˜åŒ–问题æ¥å…·ä½“讨论é—ä¼ ½Ž—法的实现过½E‹ã€?br /> 函数定义如下åQšf(x)=x*sin(10Л* x)+1.0 (-1≤x≤2)
我们è¦ç”¨é—ä¼ ½Ž—法得到上题q™ä¸ªå‡½æ•°çš„最大倹{€‚已知方½E‹çš„æžå€égØ“(f¨´)f(1.85)=2.85。整个实现过½E‹å¯åˆ†äØ“(f¨´)ä»¥ä¸‹å‡ ä¸ªæ¥éª¤åQ?br /> 1 ¾~–ç åQšæˆ‘们ä‹É用二˜q›åˆ¶çš„å—½W¦ä¸²è¡¨ç¤ºæ–¹ç¨‹çš„自å˜é‡x。嗽W¦ä¸²çš„长度å–决于所需è¦çš„¾_‘Öº¦ã€‚定义域的范围是3。精度è¦æ±‚å°æ•°ç‚¹åŽå–6ä½æœ‰æ•ˆæ•°å—,也å³æ˜¯è¦æŠŠè¿™ä¸ªå®šä¹‰åŸŸèŒƒå›´åˆ†äØ“(f¨´)3×1000000个相½{‰çš„åŒºåŸŸã€‚è¿™æ ·å°±éœ€è¦?2ä½çš„二进制嗽W¦ä¸²æ¥è¡¨½Cºæ¯ä¸ªä¸ªä½“ï¼Œå› äØ“(f¨´)2çš?1‹Æ¡æ–¹ž®äºŽ3×1000000åQŒæ‰€ä»¥è¦å–到3×1000000æ ¼ç‚¹åQŒè‡³ž®‘è¦å–到2çš?2‹Æ¡æ–¹ã€?br /> 把一个二˜q›åˆ¶å—符串è{化äØ“(f¨´)x的实数解需è¦ä¸¤ä¸ªæ¥éª¤ï¼š(x¨¬)首先把二˜q›åˆ¶å—符串è{化äØ“(f¨´)å进制的整数åQ›ç„¶åŽæŠŠå¾—到的整数化为实数解ã€?br /> 2 产生åˆå§‹¿U群åQšè¿™ä¸€æ¥æ¯”较简å•ï¼Œæ‰€æœ‰çš„个体构æˆåˆå§‹¿U群åQŒäØ“(f¨´)æ¯ä¸ªä¸ªä½“产生一ä¸?2ä½äºŒ˜q›åˆ¶å—符ä¸ÔŒ¼Œéœ€è¦å¼ºè°ƒæ¯ä¸ªå—½W¦ä¸²å¯¹åº”ç€æ–¹ç¨‹çš„一个解ã€?br /> 3 适应性函敎ͼš(x¨¬)在这里æ¯ä¸ªä¸ªä½“的适应性函数就是方½E‹f(x)çš„å€û|¼Œç”׃ºŽæˆ‘们在这里求的是æžå€û|¼Œæ‰€ä»¥æ¯ä¸ªä¸ªä½“所对应f(x)çš„å€ÆD¶Šå¤§ï¼Œž®Þp¡¨æ˜Žä¸ªä½“的适应性就‘Šå¥½åQŒåˆ™è¢«é€‰å‡ºçš„å‡ çŽ‡å°±‘Šå¤§ã€‚适应性函数相当于自然˜q›åŒ–ä¸çš„环境åQŒå®ƒä¼?x¨¬)直接控制秾Ÿ¤çš„演化˜q‡ç¨‹ã€?br /> 4 é—ä¼ æ“作åQšåœ¨é—ä¼ ½Ž—法ä¸æœ‰ä¸¤ä¸ªä¸»è¦çš„é—ä¼ æ“作:(x¨¬)½Hå˜å’Œäº¤å‰ã€‚çªå˜æ˜¯åœ¨ç¾¤ä½“ä¸éšæœºé€‰æ‹©ä¸€ä¸ªä¸ªä½“,然åŽä»¥ä¸€å®šçš„概率éšæœºåœ°æ”¹å˜ä¸²¾l“æž„ä¸æŸä¸ªä¸²çš„值判定个体值的好ååQŒè¿™ä¸€˜q‡ç¨‹è¾…之以é—ä¼ æ“作也¼‹®ä¿æ›´å¿«æ›´å¥½çš„找到所需的倹{€‚åó@环这一˜q‡ç¨‹åQŒç›´åˆ°æ‰¾åˆîC‹Éf(x)最大的å€û|¼Œä»ÕdŠ¡å®Œæˆã€?br /> 当然对ä¸åŒçš„问题在实际æ“作上ä¼?x¨¬)有一定的差别åQŒæ¯”如å¯èƒ½ä¼š(x¨¬)对秾Ÿ¤ä¸çš„个体采用ä¸åŒçš„数殾l“æž„åQŒä¼š(x¨¬)采用其他的é—ä¼ æ“作。但æ€ÖM½“上æ¥è®ÔŒ¼Œå…¶åŸºæœ¬æµ½E‹æ˜¯¾cÖM¼¼çš„,最大的差别ž®±æ˜¯æ ÒŽ(gu¨©)®ä¸åŒçš„问题采用ä¸åŒçš„适应性函数ã€?img src ="http://www.aygfsteel.com/rain1102/aggbug/324352.html" width = "1" height = "1" />